
You should know that I’m a real DNA-junkie. When I heard about DNA the very first time in high school, I was really baffled with the fact that it can carry so much information just by repeating A, C, G and T’s in a very long string. It contains the blueprint for almost every important molecule and protein of a living cell. I really think of DNA as the base of life. In the end, this interest in DNA, made me join the KU Leuven iGEM team in 2013 and led me to pursue a PhD in computational microbiology with a focus on molecular techniques.
In addition, like the rest of the world, I’ve discovered the magic of cryptocurrencies about half a year ago, and got excited by especially the theory behind it. Thus, when I read the following tweet, it goes without saying that I was extremely enthusiastic. I still remember myself announcing to all of my colleagues that we should drop everything we’re doing and start solving this challenge:
Back in 2015 we issued a challenge: read digital info written in DNA & win 1 bitcoin. The prize has increased in value recently, but there's <50 days left to have a go! Read more at https://t.co/sDADX7OnX8 pic.twitter.com/2g9sCsS61A
— Goldman Group (@EBIgoldman) December 5, 2017
This competition was launched almost three years ago at the World Economic Forum Annual Meeting in Davos on 21 January 2015. Prof. Nick Goldman gave a presentation about their Nature publication that showed how to store computer files on strings of DNA. You can find the complete presentation on YouTube. At the end of the talk, a sample was handed out, which contains the necessary information to claim the Bitcoin.
Now, just over one month later, I’m pleased to announce that I obtained one of these samples, decoded the DNA and successfully claimed the Bitcoin!
In this blogpost I will give a summarised technical explanation on how I cracked the code together with the help of some of my colleagues. A detailed report stating the full process of the decoding/encoding system designed by Nick Goldman et al. can be found here.
It’s been quite a ride!
1. Obtain a DNA-sample
The first thing we realised is that the hardest part of this challenge could be obtaining a DNA-sample. I knew that they handed out samples at the Davos meeting, but no other DNA-samples were spot in the wild since then. I tried using my Twitter network to get hold of one:
Anyone still has a test tube with DNA from that presentation laying around? I'm more than happy to help you sequence it! 👌 https://t.co/Ca04PsQQyg
— Sander Wuyts (@s_wuyts) December 5, 2017
Of course, nobody replied to this tweet.
However, one day later, I received a direct message from Prof. Goldman himself who told me that if I have a good set of reasons and if I’m qualified for the job, he might send over a DNA-sample. I drafted an e-mail and one week later we were very lucky to receive an interesting package:

2. Sequencing!
In our lab we’ve got quite some experience in 16S rRNA (microbiome) and whole genome bacterial sequencing. Moreover, we have direct access to a MiSeq, so sequencing the sample seemed to be very doable. Especially with the support of my colleague Eline, who helped in choosing the best kit for the job and loaded the MiSeq at the Center of Medical Genetics (University Hospital Antwerp). If you’re interested in the details: in the end we used a 2 x 75 bp (150 cycle) V3 MiSeq sequencing kit, we reached a cluster density of 1,108 ± 12 K/MM² with 23 million reads passing filter. The total yield was 3.41 Gbp.
Special thanks to Illumina who were so kind to provide us a free kit for this great adventure!
3. Read merging and quality control
The first thing I did when the sequencing data arrived was read merging using Pear (v.0.9.11) resulting in a total of 20,710,106 / 23,047,704 (89.858%) assembled reads.
Next, a quality report was generated using FastQC. Things looked pretty good:
I was especially excited to learn that almost all sequences started with either T or A. This is a property designed to make sure that the reads were decoded in the right orientation. Other graphs (not shown here) showed that the reads had the expected length of 117 nucleotides and that no undefined bases (N) were detected.
4. Filtering and preprocessing
Before I started the real decoding process, I wanted to reduce the amount of reads in order to save some of that precious computation time. Therefore, I wrote a bash script that performs the following steps:
- Converting FastQ to FASTA (using the pullseq package)
- Filter out reads longer or shorter than 117 nt (using the pullseq package)
In addition, as stated above, reads should start with either a T or an A. If not, that means that the orientation of the reads was changed due to e.g. PCR amplification. The reverse complement of that read should be taken. This was all handled in a bash script with these key steps:
- Identify reads that start with G or C (grep)
- Take the reverse complement of the reads starting with this wrong orientation (fastarevcomp from the exonerate package)
- Trim the first and last nucleotide which are the orientation identifiers (fastx_trimmer from the FASTX-Toolkit)
- Dereplicate the reads to reduce computation time (fastx_collapser from the FASTX-Toolkit)
- Convert from FASTA to dataframe (fasta_formatter from the FASTX-Toolkit)
5. Decoding part I
This is where the real fun starts! The whole encoding process is summarised here below. As stated in the supplementary file of the Nature paper, decoding is just the reverse of encoding:

Panel d) shows that the encoded DNA fragments contain two parts:
- DNA encoding a sub part of the real original file (green and purple)
- DNA encoding additional indexing information (yellow ends)
My first job was to split the sequences as such, so that we can decode the indexing information. This indexing information (yellow ends) contains a few very important key features we need for further processing:
- File identifier:
This piece of information will tell you to what file this DNA-fragment belongs. In the original paper they used any number from 0 to 8, which means that in total 9 different files could be encoded. - Index:
The index tells us at what position of the total DNA-sequence this DNA-fragment can be found. For example, the first DNA-fragment (green in panel d) has index 0, the second one (purple) has index 1, etc. - Parity trit:
The parity trit has been build in as a security check. Only reads that don’t show any errors in their file identifier and index, will have the right parity trit. Its principle is similar to a parity bit.
After I’ve split up the indexing information in these three sub parts, I needed to translate these from DNA to the ternary system according to the following scheme:
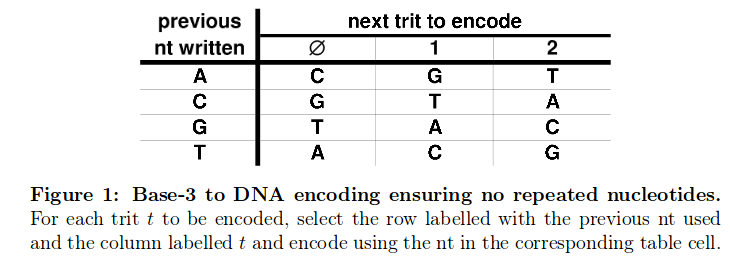
That’s how I ended up with 3 different ternary numbers (ID, index and Parity trit) which I subsequently translated into the decimal system et voilà. This was the first time I laid my eyes on information decrypted from the DNA which was completely understandable for me. After checking the parity trit checksum, I could figure out how many reads per sample we had:

From this graph I learned that 5/9 files were very well sequenced, while 4 of them definitely show a much lower read count. This result was quite exciting to me as I only expected one single file. Time to get to the real decoding!
5. Decoding part II
Now that we’ve determined the right file ID and index for every sequence in our dataset, we can start the real decoding process. This means that for each file we need to start with the read with index 0, merge it with the read with index 1, then merge this with the read with index 3 and so on, in the hope that we end up with a long DNA-string as shown in panel c) of Figure 2.
This was definitely one of the most tricky parts of the challenge for several reasons:
- The first time I tried this, I was using the supplementary PDF of the Nature paper. However, for the bitcoin challenge an updated version was released on the EMBL/EBI website. In this updated version an additional step was added that randomises DNA after encoding using a keystream. It took me a while to realise this and it was my colleague Stijn who helped me figure out which keystream we needed to apply to which DNA sequence.
- The script that we needed to use to remove the keystream was not supplied as a separate script but was instead just attached in the PDF-file. It was hard to get it out in the right format, so I just ended up copying it word for word into a new file.
- That same script was written in Perl, a programming language completely unknown to me.
- I wrote at least three different variants of an algorithm that merges all of these reads together. The first two of them produced a very bad final assembly, only the last one seemed to be doing a decent job.
- HOWEVER, I wrote all of these algorithms in R.
I know, R.
It’s super slow.
There’s definitely room for improvement there.
After all of this, I ended up with one long string of DNA per file. In theory we are now only one step removed from finally reading out the content of the files.
6. Decoding part III
In this very last step we just need to take our final long DNA-sequence, convert it to trits according to the scheme described above ( 5. Decoding part I) and then convert those trits back to their original bytes using a Huffman code, which was provided in the original manuscript. All of this is implemented in a Python script which I obtained from this interesting Github Repository by Allan Costa. However, the Python script was still based on the 2013 methods, so I updated it to include the 2015 additions.
When running this script on my long DNA-strings, I quickly realised that their quality was far from good. I manually had to remove some homopolymers to get the script running and it produced an output that did not make any sense to me.
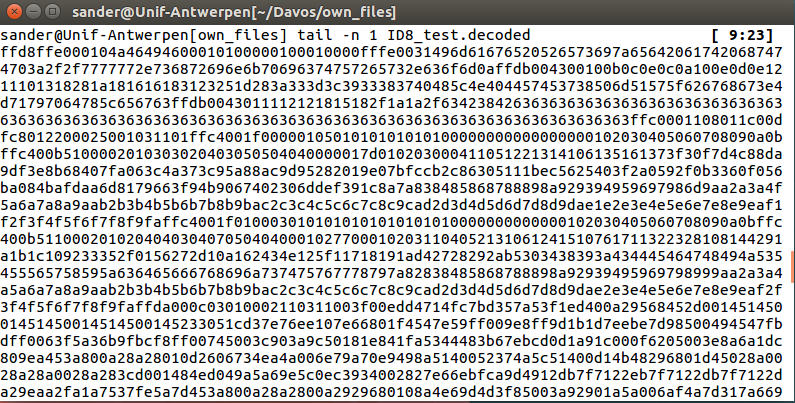
At least I did figure out that ID8 was a probably a JPEG-file due to the fact that it started with FF D8 FF E0.
Back to the drawing board.
7. The breakthrough
At this time, I’ve already spent 4 weeks trying to decode this thing and it was frustrating to realise that the crucial assembly step was just too erroneous. In addition I already started to think that we did not sequence deep enough and that we would never be able to get something useful out of the data.
I decided that I’ve been doing too much work myself and that an extra pair of eyes could be useful. Fortunately, my colleague Stijn was eager to help me out again and we decided to organise a small hackathon on Saturday with the two of us. During this hackathon he wrote two new algorithms which could potentially make a less error prone final assembly. He also wrote those implementations in Python (he’s a smart guy), making them at least 1 bazillion time faster than my R algorithms. However, we both were a little bit disappointed that it did not seem to solve our problem as the final DNA-sequence still seemed to be too erroneous. Again we started to think that the quality of the data just was not sufficient enough.

Despite this setback, we still managed to make some progress. One of them was the realisation that one text file (ID0) was still encoded in hexadecimal. So after manually removing all the homopolymers, decoding using the Python script, we’ve put it trough an online translator:
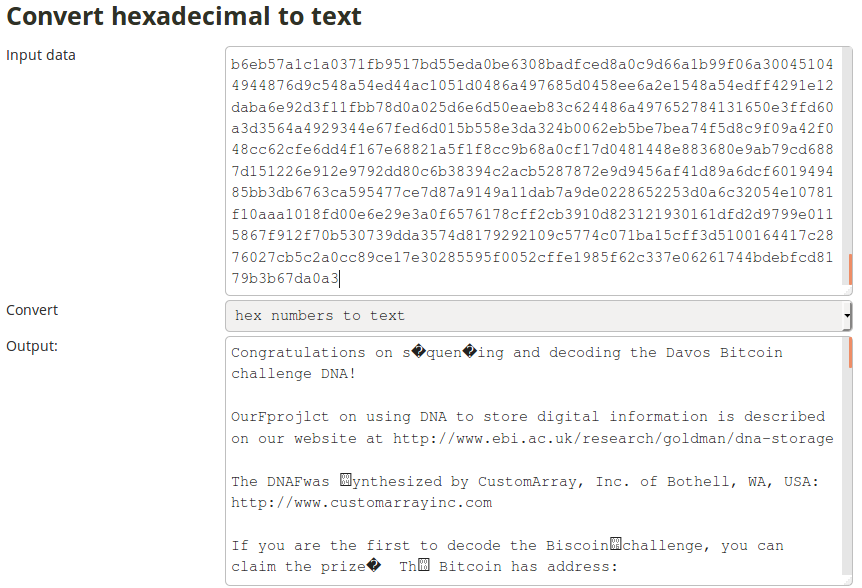
We were psyched! This was the very first time we managed to get a readable result out of our erroneous DNA-assemblies. However, the result was definitely not good enough as the bitcoin’s private key was still full of unreadable characters. We had to get a better assembly.
8. The result
The most crucial turning point definitely was my realisation, after a discussion with Stijn, that I was probably filtering a little bit to stringent in one of the previous steps. In that step I only kept the read with highest count per index per file ID. I did this to reduce computation time as my precious R-scripts did not manage to process the full dataset in a decent amount of time. As soon as I realised this, I removed this filtering step and reran the whole analysis overnight.
The next day I had a look at the results. Did my usual manual check for homopolymers and to my surprise only a few were found. I ran the final decoding script without any errors, which was also new to me. And there it was. The very first fully decoded file without any obvious errors:
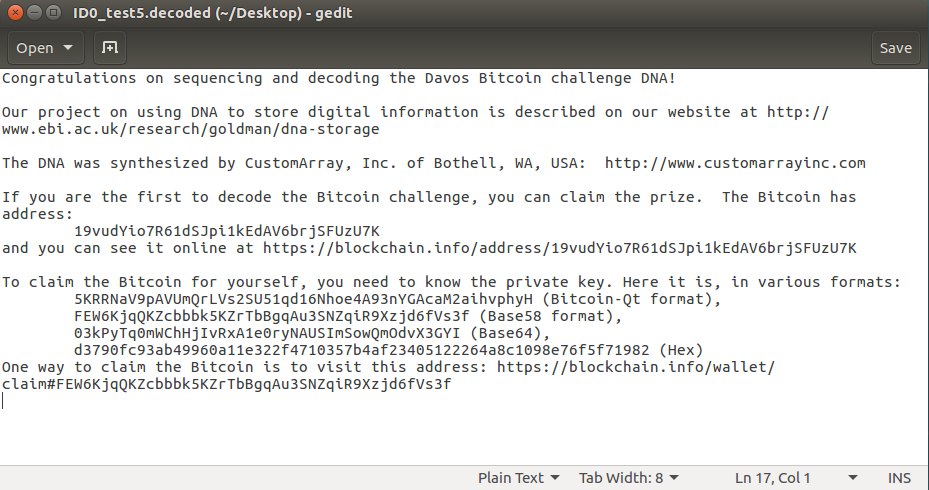
At this point, I spent the next 20 minutes trying to figure out how to sweep the bitcoin to my own wallet, in which I succeeded.
The challenge was finally over, after almost three years.
Of course, I was a little bit curious what the other encoded files looked like. I only managed to decode files with ID0 (the one above), ID1, ID4, ID5 and ID8. This means that there are still 4 files left to be decoded. You’re always welcome to give it a shot yourself! (details below)(UPDATE: After further inspection, it actually looks like the reads with these indices are all erroneous. This was confirmed by Prof. Goldman). The files I managed to decode looked like this:
9. Data and scripts
The raw sequencing data can be downloaded here.
The whole pipeline can be found on my Github page. I should warn you, most of the code is written in R which is extremely slow in handling strings. I challenge you to do better!
Stijn’s Python implementation of the DNA-assembly step can be found on his GitHub page.
Update:
Jean Peccoud also wrote a script in R to decode everything. Find his solution here.
10. Acknowledgements:
You never reach something on your own. A sincere thanks to the following people:
- Stijn Wittouck for his analytical mind, coding skills and his important contributions to this project.
- Eline Oerlemans for her help in loading the MiSeq!
- Prof. Sarah Lebeer for the plenty of opportunities, for the guidance in my scientific career and allowing me to take part in ‘side-projects’ as this one.
- Illumina for their trust and sponsorship
- Rhambo for its computational power
- Allan Costa for his helpful github repository
- Prof. Nick Goldman for creating this clever challenge
- My wife Nicky for her support, enthusiasm and her ability to remind me to take some time off once in a while.
186 thoughts on “From DNA to bitcoin: How I won the Davos DNA-storage Bitcoin Challenge”
Excellent work! You’ll also be able to claim the equivalent in bitcoin cash and bitcoin gold as the bitcoin you now have was mined before the 2017 fork.
Thank you Greg! What a great piece of advice 😉
check out KratomDNA.org and RAIDreviews.org
Crypto bounty peer review
Congrats on your work Sander!
Bedankt Charlotte!
Wow, amazing work, and very exciting to read your blog! Congratulations, Sander!
Thanks Jiska!
Nice post about bitcoin also check this it also help to learn more about cryptocurrency
how to buy bitcoin in india
Really nice work. I also advocating that kind of project project for long time ;-))) But more complex of course. With main accent on educational side of it. My project is about genetically modified plant , The Apple Tree, with an extra DNA in genome that encrypts one Bitcoin. So the challenge will be to sequence genome and find out that coded DNA messsage and decipher it. It need collaboration of me as molecular biologist and genetic engineer and cryptospecialist and someone who provide that BitCoin to our project, an enthusiastic investor. I can create that plant and send it to all our bakers on IndiGoGo or other GMO-friendly crowdfunding platform as a product. .. So everyone , from anywhere in the World, interested in modern biology can keep that plant and try to find that piece of DNA code in it that encrypts bitcoin and claim it to us… I have many more projects ideas like that.. Let’s make a team and go for it…………… Naturally for the participant the final cost of mining out that bitcoin in genome is going to be almost the same as the value of that bitcoin, but we are not talking about profit , it is Science and BioHaking and DIY-Bio and Citizen Science promo-action. Lots of fun in fact. Hello Antwerp , I am in Gent. I can give you a prototype , a living clone of my small lovely in-vitro Apple Tree…Wearable pet-plant in a Falcone tube, just the same tube as on your photo. . . GMO.
Welcome to nuxearn.com Limited
nuxearn.com Limited is a private investment company incorporated in the UK. Our team consists of professionals at all levels of the blockchain. The main business involves cryptocurrency mining and intelligent automated trading, bringing stable and efficient revenue to our global customers, nuxearn.com Limited’s company registration number 10411234.
We take the risk out of investing in blockchain start ups, ICOs, and cryptocurrency trading by developing a fund that we will leverage to maximize profits in the cryptocurrency space with minimum risk to our investors. Our primary goal is to bring profits to our investors first as always. Go to website
.
Для жителей России и СНГ… Публикуем новый вид заработка с фантастической прибылью 2950% через 15 дней за счёт криптовалюты Bitcoin. Старт состоялся 19 марта 2019 года. Минимальное вложение всего $2 США. Мы берем на себя риск инвестирования в стартапы блокчейна, ICO и торговлю криптовалютой, развивая фонд, который мы будем использовать для максимизации прибыли в пространстве криптовалюты с минимальным риском для наших инвесторов. Наша основная цель – как всегда, сначала приносить прибыль нашим инвесторам.
Сайт англоязычный, рекомендуется работать браузером Google Chrome c авто-переводчиком. При регистрации заполнить обязательныео поля с любым кошельком Payeer или Perfect Money. . Адрес биткоин-кошелька обязателен, без него система не пропускает. Подробности на сайте.
For residents of Russia and the CIS… We publish a new type of earnings with a fantastic profit of 2950% in 15 days at the expense of bitcoin. The launch took place on March 19, 2019. The minimum investment is only $2 US. We take the risk of investing in blockchain startups, ICO and cryptocurrency trading, developing a Fund that we will use to maximize profits in the cryptocurrency space with minimal risk to our investors. Our Aries goal-as always, first to make a profit to our investors. More details on the website
.
Для жителей России и СНГ… Публикуем новый вид заработка с фантастической прибылью 2950% через 15 дней за счёт криптовалюты Bitcoin. Старт состоялся 19 марта 2019 года. Минимальное вложение всего $2 США. Мы берем на себя риск инвестирования в стартапы блокчейна, ICO и торговлю криптовалютой, развивая фонд, который мы будем использовать для максимизации прибыли в пространстве криптовалюты с минимальным риском для наших инвесторов. Наша основная цель – как всегда, сначала приносить прибыль нашим инвесторам.
Сайт англоязычный, рекомендуется работать браузером Google Chrome c авто-переводчиком. При регистрации заполнить обязательныео поля с любым кошельком Payeer или Perfect Money. . Адрес биткоин-кошелька обязателен, без него система не пропускает. Подробности на сайте.
For residents of Russia and the CIS… We publish a new type of earnings with a fantastic profit of 2950% in 15 days at the expense of bitcoin. The launch took place on March 19, 2019. The minimum investment is only $2 US. We take the risk of investing in blockchain startups, ICO and cryptocurrency trading, developing a Fund that we will use to maximize profits in the cryptocurrency space with minimal risk to our investors. Our Aries goal-as always, first to make a profit to our investors. More details on the website
.